Étude FIRST
Fiche descriptive de l'étude
Étude ovaire
FIRST
Titre de l'étude
The FIRST (First line ovarian cancer treatment with Niraparib plus TSR 042) Study: Essai randomisé adaptatif de phase III comparant un traitement standard à base de platine avec du platine associé au TSR-042 suivi de l’association TSR-042 et Niraparib puis de TSR-042 en thérapie de maintenance, chez des patientes atteintes d’un cancer épithélial ovarien, des trompe de Fallope ou péritonéal de stade III ou IV.
Statut
Recrutement terminé / Suivi en cours
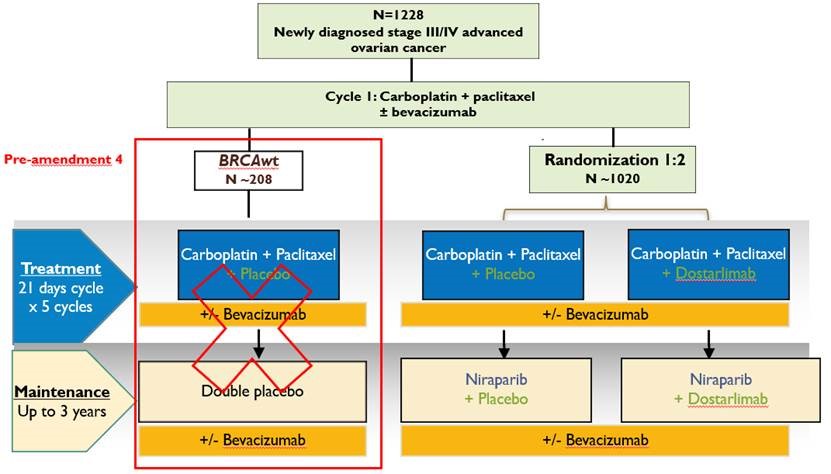
Plan de traitement de l'étude
Promoteur
Nom du promoteur : TESARO / GSK
Groupe Leader : GINECO
But
Objectif primaire
Comparer la survie sans progression.
Objectifs secondaires
- PFS selon irRECIST
- Survie globale
- Maintien de la PFS selon irRECIST
- Qualité de vie
- Taux de réponse complète pathologique (pCR) chez les patientes ayant subit une chirurgie de cytoréduction intervallaire
Objectif exploratoire
Evaluer la pharmacocinétique et l’immunogénicité du TS-042.
Phase
Phase III
Type de patiente
Cancer ovarien non-mucineux inopérable de stade III ou IV, dont une chimiothérapie néo-adjuvante est envisagée, ou une maladie avec résidu macroscopique après une chirurgie primaire de cytoréduction.
Nombre de patientes récrutées
1335 patientes randomisées dont 340 patientes en France.
Principaux critères d'inclusion
- Patiente présentant un cancer épithélial ovarien non-mucineux de haut grade
- Tous les stades IV sont éligibles
- Les patientes de stade III sont éligibles si elles correspondent aux critères suivants :
- Les stades IIIC avec une cytoréduction complète (CC0), soit avec un résidu maximal de 5 cm au total
- Tous les stades III inoperables ou avec un résidu tumoral macroscopique après une chirurgie primaire de cytoréduction
- Tous les stade III avec un résidu tumoral macroscopique après une une chirurgie primaire de cytoréduction
- Tous les stades III doivent avoir une chimiothérapie néoadjuvante plannifiée
- Patiente acceptant de fournir un échantillon de sang et de tumeur à l’inclusion et de compléter des questionnaires de qualité de vie
Principaux critères de non-inclusion
- Patiente atteinte d’un cancer mucineux, des cellules germinales, des cellules transitionnelles, et bas grade ou tumeur indiférenciée.
- Patiente de stade III avec une résection RO après une chirurgie primaire de cytoréduction
- Patiente n’ayant pas correctement récupéré de sa chirurgie primaire • Patiente ayant reçu une thérapie antérieure d’anti-mort cellulaire programmé 1 (PD-1) ou d’anti ligand mort cellulaire programmé 1 (PDL-1) ou d’inhibiteur de PARP.
- Patiente immunodéprimée
Bilans
Baseline
- Examen clinique, biologique, ECG
- Scanner ou IRM
- Prélèvement sanguin pour le test ADNct HRR nécessaire pour la randomisation
- Questionnaires de qualité de vie EORTC-QLQ-C30, EORTCQLQ- OV28, and EQ-5D-5
Période de traitement
- Chimiothérapie A chaque cycle
- Examen clinique et biologique
- Ca 125
- Questionnaires de qualité de vie EORTC-QLQ-C30, EORTCQLQ- OV28, and EQ-5D-5 (aux cycles 1, 2, 4 et 6)
- Au cycle 4
- Scanner ou IRM
- Ca 125
- Maintenance A chaque cycle
- Examen clinique et biologique
- Ca 125
- Questionnaires de qualité de vie EORTC-QLQ-C30, EORTCQLQ- OV28, and EQ-5D-5 (Tous les cycles durant les 3 premiers cycles puis tous les 3 cycles jusqu’au 18ème cycle, puis tous les 6 cycles jusqu’à progression). Tous les 4 mois jusqu’à 24 mois, puis tous les 6 mois jusqu’à 36 mois, tous les an au-delà de 36 mois.
- Scanner ou IRM
- Ca 125
Visite de fin de traitement
Juste après l’arrêt du traitement :
- Examen clinique et biologique
- IRM ou scanner
- Ca 125
- Questionnaires de qualité de vie EORTC-QLQ-C30, EORTCQLQ- OV28, and EQ‑5D-5 Safety
Follow-Up
30 jours après l’arrêt du traitement
- Examen clinique et biologique
- IRM ou scanner
- Ca 125
Retour
Accés pro