Étude UTOLA
Recrutement terminé / suivi
Fiche descriptive de l'étude
Étude ovaire
UTOLA
Titre de l'étude
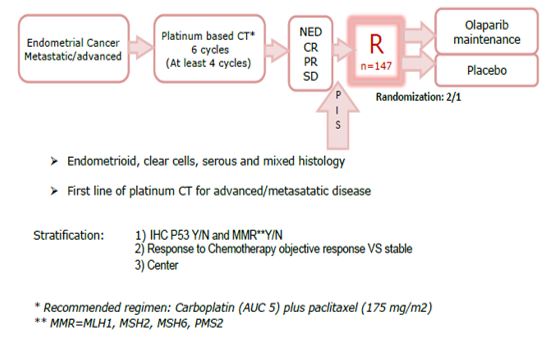
UTOLA : Etude de phase II randomisée en double aveugle contre placebo, multicentrique évaluant l’Olaparib comme traitement de maintenance chez des patientes sensibles au platine pour un carcinome de l’endomètre avancé ou métastatique.
Statut
Recrutement terminé / suivi
Plan de traitement de l'étude
Promoteur
ARCAGY - GINECO
But
Objectif Principal
Déterminer l’efficacité d’une maintenance par Olaparib chez des patientes sensibles au platine avec un carcinome avancé ou métastatique de l’endomètre en évaluant la PFS1 selon RECIST
Objectifs Secondaires
- La survie globale (OS)
- Le taux de réponses objectives (ORR)
- Déterminer la PFS1 et le taux de réponse objective (ORR) selon les statuts IHC P53 et MMR et NGS BRCA/HRD
- Déterminer le temps entre la randomisation et l’initiation de la première et seconde thérapie.
Phase
Phase II
Type de patiente
Patientes ayant un cancer de l’endomètre avancé ou métastatique
Nombre de patientes recrutées
147 patientes dans 36 centres
Critère principal d’évaluation
- Date de progression PFS (RECIST) et Date de seconde progression (PFS2)
- Profil moléculaire : MMR/MSI et P53
- Date de décès : Survie globale
- Evènements indésirables
- Questionnaires qualité de vie
Haut de page
Modifié le
05-10-2021 11:06:49
Retour
Actu
Accés pro
Accés pro
Accès professionnel
Actualités






Le GINECO à l'ESGO 2024 à Barcelone
Le GINECO à l'ESMO 2023
Saint Paul 2023
Nouvelle étude internationale : SALVOVAR !
Application Mobile Essais Cliniques
Congrès Trophoblasique génitale