Étude BOLD
Fiche descriptive de l'étude
Étude ovaire
BOLD
Titre de l'étude
BOLD : Etude de phase II évaluant la tolérance et l'efficacité de l'association Bevacizumab (FKB238), Olaparib et Durvalumab (MEDI 4736) chez des patientes atteintes d'un cancer de l'ovaire épithélial avancé en rechute
Statut
Le recrutement de cette étude est terminé.
Plan de traitement de l'étude BOLD
Promoteur
ARCAGY-GINECO
But
Objectif primaire
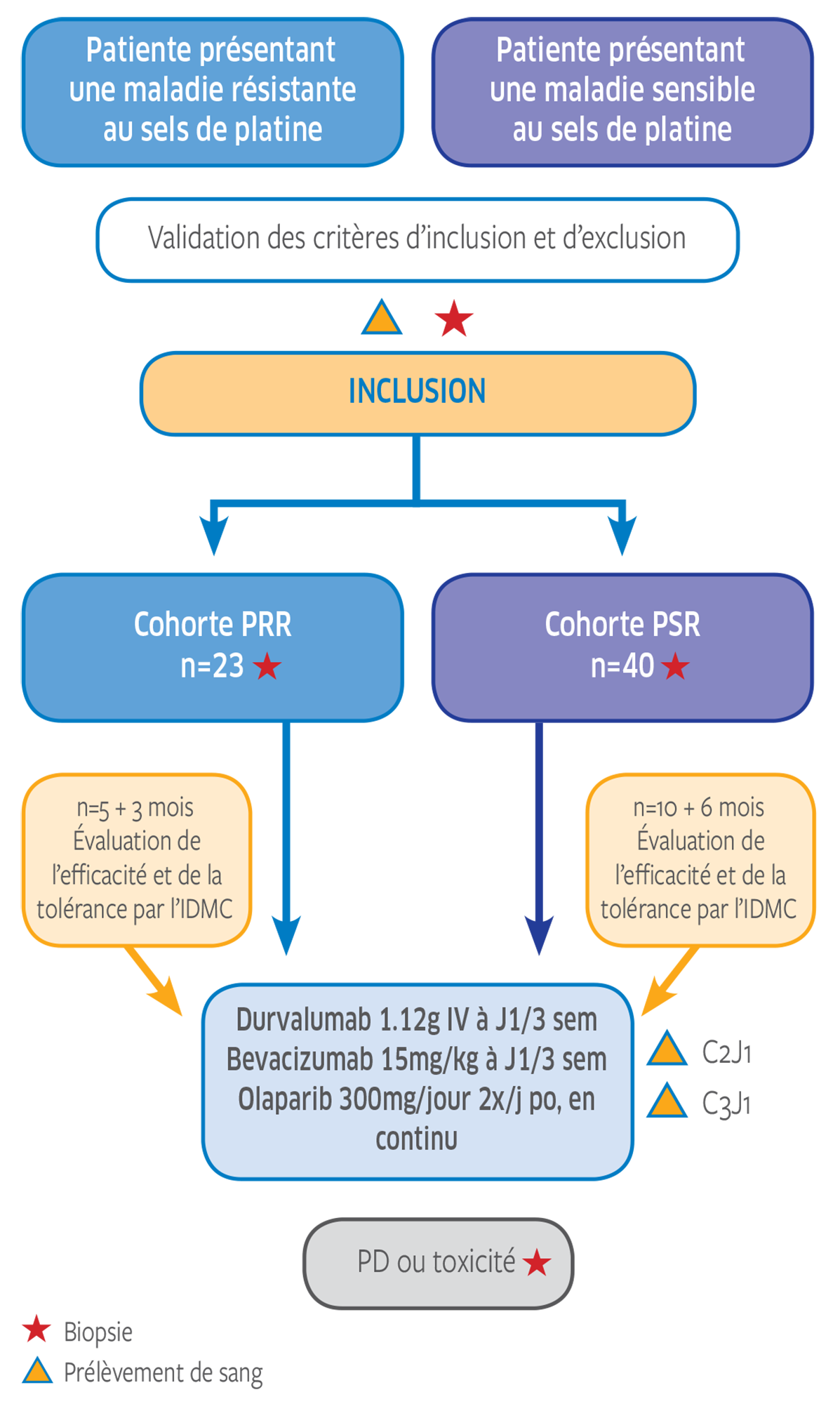
Efficacité et tolérance de l’association suivante:
Durvalumab 1.12 g IV Q3W
Bevacizumab 15 mg/kg Jour 1 Q3W
Olaparib 300 mg deux fois par jour, p.o. (comprimés de 150 mg et 100 mg), en continu (NB : avec l’utilisation de la nouvelle formulation sous forme de comprimés, la dose de 300mg est considéré comme équivalente aux capsules de 400mg)
L’objectif primaire est le taux de non-progression clinque et radiologique de la maladie, selon les critères de réponses liés au système immunitaire (irRC) (Wolchok et al. 2009) : et al. 2009) :
- A 3 mois pour la cohorte RRP
- A 6 mois pour la cohorte RPS
Objectifs secondaires
Les objectifs secondaires sont:
- L’analyse de la baisse du CA 125 selon le paramètre KELIM
- La survie sans progression (SSP)
- La survie globale (SG)
- La réponse tumorale
- La toxicité selon l’échelle CTCAE V.5.0
Objectifs de recherche translationnelle
Les objectifs de recherche translationnelle sont:
- De corréler l’administration de l’olaparib et l’efficacité du durvalumab
- De corréler le phenotype HRD avec la réponse à la théraoie anti-PARP
- De corréler le microenvironnement tumoral, le statut immunitaire et la réponse au Durvalumab
Phase
Phase II
Type de patiente
- Patiente présentant un cancer de l'ovaire, péritonéal primitif et / ou cancer des trompes de Fallope, histologiquement confirmé (d'après les résultats histopathologiques locaux) : tumeur ovarienne séreuse de haute grade ou endometrioïde de haut grade ou autre tumeur épithéliale non mucineuse de haut grade.
- Patiente ayant reçu au moins une ligne de chimiothérapie par platine-taxane et présentant une rechute :
- Soit résistante au platine (<6mois après la dernière dose de platine)
- Soit sensible au platine (≥ 6mois après la dernière dose de platine)
- Patiente n'ayant reçu aucun des médicaments testés, ou ayant précédemment reçu soit du bevacizumab soit de l'Olaparib MAIS PAS la combinaison des deux médicaments.
- Présence d’au moins une lésion mesurable ou évaluable et permettant une évaluation répétée selon irRC. L'analyse de base doit être obtenue dans les 28 jours suivant la première dose.
- Disponibilité d'un échantillon tumoral prétraitement (bloc FFPE archivé ou biopsie fraîche si possible) réalisé 3 mois ou moins avant l'inclusion dans l'étude et réalisée APRÈS la dernière administration de chimiothérapie.
Nombre de patientes recrutées
74 patientes sont incluses dans 9 centres.
Critère principal d’évaluation
Calcul du nombre de patientes nécessaire et analyse de l’obectif primaire
Les considérations statistiques sont indépendantes dans les deux cohortes. La taille de l'échantillon a été calculée selon une distribution binomiale exacte, en utilisant un plan en une étape :
Cohorte résistante au platine
Un taux de maladie non progressive de 50% à 3 mois est considéré comme indésirable (p0 = 50%) par rapport au contrôle historique dans cette population de patientes (Pujade-Lauraine et al, 2014). Un taux de maladie non progressive de 75% ou plus (p1 = 75%) est considéré comme méritant des invsetigations complémentaires.. Vingt-trois patientes seront incluses dans l'essai (n = 23) avec un risque d’erreur unilatéral de type 1 : α = 5% et une puissance de 80% pour un taux observé de maladie non progressive de 75%. L'hypothèse nulle (p ≤ p0) sera rejetée si la limite inférieure de l'intervalle de confiance bilatéral à 90% est supérieure à 65%.
Cohorte sensible au platine
Un taux de maladie non progressive de 65% à 6 mois est considéré comme indésirable (p0 = 65%) par rapport au contrôle historique dans cette population de patientes (Pujade-Lauraine et al, 2011). Un taux de maladie non progressive de 84% méritera des investigations supplémentaires (p1 = 84%). Quarante patientes seront incluses dans l'essai (n1 = 40) pour un taux d'erreur unilatéral de type 1 : α = 3% et une puissance de 82% lorsque le taux observé de maladie non progressive est de 84%. L'hypothèse nulle sera rejetée si la limite inférieure de l'intervalle de confiance bilatéral à 90% est supérieure à 65%.
Retour
Accés pro