
Gliomes
Ce sont les tumeurs cérébrales primitives les plus fréquentes...
LEUR POINT DE DEPART : L’ECTODERME
A l’origine, l’embryon est constitué de trois feuillets embryonnaires primitifs, l’endoderme, le mésoderme et l’ectoderme.
L’ectoderme est le feuillet externe. Sa différenciation, durant le développement de l’embryon donnera naissance à la peau et ses annexes d'une part et au système nerveux, d'autre part.
LES CARACTERISTIQUES DE CES TUMEURS
Une prolifération à partir des cellules de la glie (tissu de soutien des neurone)
Les gliomes proviennent des cellules contenues dans le tissu de soutien du cerveau, la glie, d'où le terme générique de gliome. En fait, la tumeur se développerait à partir de cellules neurales qui auraient acquis un potentiel tumoral à la suite de mutations.
Ils se développent à partir de petites cellules en forme d'étoiles appelées astrocytes. Les astrocytes contribuent à la nutrition du système nerveux central.
Les gliomes sont des tumeurs qui infiltrent progressivement le parenchyme cérébral, causant effet de masse et perte neuronale.
Les astrocytomes constituent près des deux tiers de toutes les tumeurs primitives du système nerveux central. L’âge moyen de découverte, chez l'adulte, est de 56 ans.
La tumeur primitive du cerveau la plus commune chez l'adulte
Avec une prévalence d’environ 5 à 8 cas pour 100 000 habitants et par an, les gliomes sont les plus fréquentes des tumeurs cérébrales primitives de l’adulte. Leur incidence augmente. L’exposition au champ magnétique de basse fréquence est un facteur favorisant démontré.
De 2 500 à 3 000 nouveaux cas sont diagnostiqués, par an, en France. Les hommes sont plus fréquemment touchés.
La plupart des cas sont sporadiques. Dans de rares cas, ils sont associés à certains cancers familiaux ((neurofibromatose de type I, sclérose tubéreuse de Bourneville, syndrome de Turcot, syndrome de Li–Fraumeni et syndrome de Lynch).
Environ 75 % des gliomes diagnostiqués sont de haut grade (III ou IV de la classification de l’OMS).
Leur localisation dans le système nerveux
Les gliomes peuvent se développer dans n'importe quelle région du cerveau ou de la moelle épinière. Ils infiltrent progressivement le parenchyme cérébral et causent, ainsi, un effet de masse.
Chez l’adulte, les astrocytomes se développent le plus souvent dans les hémisphères cérébraux et plus précisément dans le lobe frontal ou le lobe temporal.
Chez l'enfant, ils se développent plus volontiers au niveau de la partie du cerveau la plus basse et qui est située dans la fosse postérieure (au-dessous d'une membrane – méninge – appelée la tente du cervelet).
Les gliomes du tronc cérébral sont le plus souvent des astrocytomes de faible grade. Ils posent des problèmes spécifiques, car le tronc cérébral contrôle beaucoup de fonctions vitales. De ce fait, le plus souvent, ces tumeurs ne peuvent pas être enlevées chirurgicalement.
Le principe des différentes classifications
LE GRADE HISTOLOGIQUE
Le grade de malignité est déterminé par le degré de différenciation de la tumeur et sa cellularité, ainsi que la présence d’atypies cyto-nucléaires, d’activité mitotique, de prolifération micro-vasculaire et de nécrose.
Les tumeurs gliales sont des tumeurs histologiquement hétérogènes, dérivées des différentes cellules de la glie.
La classification anatomo-pathologique de référence actuelle est fondée sur les critères de l’OMS de 2007. Elle distingue les gliomes en fonction de leur morphologie et de leur degré de malignité croissant (grade histopronostique côté de I à IV).
VERS UNE CLASSIFICATION BIOMOLÉCULAIRE...
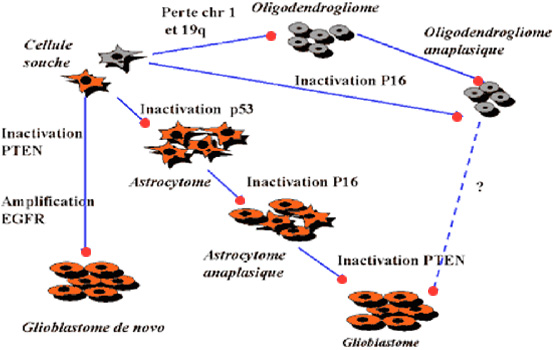
Des équipes indépendantes de chercheurs sont parvenus, grâce à l'utilisation de puces génomique, à isoler des mutations fréquentes affectant les gènes : IDH1, TP53, ATRX, CIC, FUBP1, NOTCH1 et le promoteur TERT. A partir de ces données, les chercheurs pensent qu'il existe au moins quatre grands sous-types génétiques de gliomes :
- Les gliomes de grade II et III avec co-délétion chromosomale 1p19q possèdent dans tous les cas une mutation associée du gène IDH1 ou IDH2 et sans co-délétion 1p19q mais avec une mutation du gène IDH1 ou IDH2 , qui est souvent associée à des mutations du gène P53
- Les glioblastomes avec mutation du gène IDH1 ou IDH2 sont le plus souvent des glioblastomes secondaires à des astrocytomes de bas grade et sans mutation du gène IDH1 sont des glioblastomes primitifs (de novo)
Classification des gliomes en fonction du grade
| Grade | Les différents gliomes selon le grade histologique |
|---|---|
| Grade I |
|
| Grade II |
|
| Grade III (anaplasique) |
|
| Grade IV |
|
L’astrocytome pilocytique de grade I
C'est une tumeur à développement lent, a priori bénigne.
Elle est principalement rencontrée chez l’enfant, chez qui elle représente environ 15 à 20 % des tumeurs cérébrales primitives, plus rarement chez le jeune adulte.
Elle est, le plus souvent, sous tentorielle (sous la faux du cervelet), elle rend compte de 85 % des astrocytomes cérébelleux.
Cette tumeur présente de fréquentes altérations des gènes de la famille RAF ( BRAF et RAF 1).
Ce sont des tumeurs plutôt de bon pronostic. Une résection neurochirurgicale complète de la tumeur permet une guérison définitive.
Les astrocytomes de Grade II et III
ASTROCYTOME DE GRADE II
Les principales caractéristiques
Ce type tumeur représente environ 20 % des gliomes.
Il est plus fréquemment diagnostiqué chez les sujets jeunes. L’âge moyen de survenue est en moyenne de 34 ans et peut se voir entre 20 et 45 ans.
Il existe une forte prédominance masculine.
Il peut rester focal, c'est-à-dire circonscrit à une région délimitée du cerveau ou être infiltrant.
Il est retrouvé plus volontiers dans le lobe frontal ou le lobe temporal. Il se développe dans la substance blanche, parfois cortex. Les cellules tumorales migrent le long des fibres de la substance blanche.
La tumeur peut être longtemps asymptomatique. Chez deux tiers des patients, le gliome est découvert à la suite de crises d’épilepsie.
L’IRM est l’examen de référence qui montre une lésion généralement hypo-intense en T1 et hyper-intense en T2 ou en FLAIR, siégeant volontiers dans les régions frontale, temporale ou insulaire.
Il n’existe généralement pas ou peu de prise de contraste. Dans le cas contraire, celle-ci doit faire fortement suspecter une transformation anaplasique.
Le pronostic
Leurs principaux facteurs pronostiques sont, l'âge (bon pronostic avant 50 ans), l'état fonctionnel au moment du diagnostic, leur localisation dans le cerveau (profonde, frontale) et l'existence d’un syndrome de masse.
C’est une tumeur à développement lent. Cependant, non traité, plus d’un astrocytome sur deux de grade II évolue vers le grade III ou le grade IV dans un délai qui varie de 1 à 10 ans ; en moyenne de 4 à 5 ans.
ASTROCYTOME DE GRADE III
Les principales caractéristiques
Cet astrocytome est quelquefois appelé astrocytome anaplasique. Ils sont souvent issus d'une transformation d'un gliome de grade II.
Il représente le tiers des astrocytomes diagnostiqués.
Son incidence annuelle est de 3 à 4 cas pour 100 000 habitants.
L’âge de survenue se situe entre 40 et 50 ans. Il y a une forte dominance masculine.
Cette tumeur est localisée dans la substance blanche, plus rarement dans le cortex cérébral.
L'IRM est le standard actuel pour le diagnostic et le suivi de ces tumeurs.
Ces tumeurs ont des comportements biologiques et cliniques différents, selon leur statut mutationnel de gènes, IDH1 et IDH2, codant pour deux isoformes de l’enzyme isocitrate déshydrogénase (IDH). De ce fait, les astrocytomes sont maintenant classés en : astrocytome anaplasique, IDH-muté, III Astrocytome anaplasique, IDH-non muté et NOS.
Les grandes lignes du traitement
La neurochirurgie à trois objectifs :
- Diagnostic : pour définir la suite du traitement et éliminer avec certitude un diagnostic différentiel potentiellement curable
- Thérapeutique dans le but d'améliorer le pronostic et/ ou soulager des symptômes par une réduction du volume tumoral, évacuation d'un contenu kystique, la libération ou dérivation du liquide céphalo-rachidien
- Potentialisateur es traitements adjuvants par une exérèse des zones peu vascularisées donc plus résistantes aux chimiothérapie et à la radiothérapie
La radiothérapie isolée ou suivie d’une chimiothérapie est le traitement postopératoire standard. Le gamma-knife peut être discuté au cas par cas. Dans les semaines qui suivent, une aggravation transitoire peut être observée (œdème). Une corticothérapie peut alors être utile.
Les molécules ayant démontré une efficacité sont : BICNU, CCNU (lomustine) , fotémustine, cisplatine, carboplatine, procarbazine, étoposide, cyclophosphamide, hydroxy-urée, bléomycine, témozolomide en première ligne). Des implants de carmustine peuvent également être utilisés.
Le pronostic
Il est réservé.
L’astrocytome de grade IV ou glioblastome
L'ÈPIDEMIOLOGIE
Ce sont les tumeurs cérébrales primitives les plus fréquentes chez l’adulte (60 à 70% des gliomes) et les plus invasives. C'est que l'on appelle le "cancer du cerveau".
Les glioblastomes sont environ 2 à 4 fois plus fréquents que les astrocytomes de grade III (anaplasiques) et représentent le quart des tumeurs cérébrales primitives de l’adulte.
L’incidence annuelle est de 3 cas pour 100 000 habitants, soit, en France, environ 3 500 nouveaux cas. Son incidence est en constante augmentation depuis 1990 dans les deux sexes. Il représente environ 60 % des tumeurs du SNC et prédomine chez l’homme (rapport hommes/femmes égal à 1,6).
L'âge moyen de découverte est de 65 ans. Elle atteint un maximum entre 70 et 80 ans avec 33 cas pour 100 000 habitants par an. En 2015, environ 50 % des nouveaux cas de glioblastomes sont diagnostiqués chez des patients de plus de 65 ans.
LES FACTEURS DE RISQUE
Constitutionnels
Environ 5 % des glioblastomes ont une histoire familiale en dehors de tout syndrome identifié. Dans 1 % des cas, il s’agit d’un syndrome génétique identifié comme le syndrome de Li-Fraumeni, le syndrome de Turcot ou la neurofibromatose de type 1 ou 2.
Des variations génétiques fréquentes de 7 gènes augmenteraient le risque de gliome .
Il se voit surtout chez l'homme.
Environnementaux
Il n’y a pas de lien prouvé avec l’alimentation, le tabac, l’ingestion de composés nitrosés ou les traumatismes cérébraux.
Seule l’exposition aux radiations ionisantes semble augmenter le risque de gliomes. Les données concernant l’utilisation des téléphones portables sont contradictoires.
LOCALISATION & EVOLUTION
Le glioblastome se développe dans l'un des deux hémisphères cérébraux et augmente rapidement de volume.
Il s'accompagne d'un œdème important et se traduit par des signes neurologiques dus à la lésion des cellules nerveuses (paralysie, troubles sensitifs) et à une hypertension intracrânienne se traduisant par des maux de tête, surtout le matin.
DEUX TYPES DE GLIOBLASTOMES
Des caractéristiques différentes
La tumeur peut survenir de novo ou résulter de l’évolution d’un astrocytome de bas grade
Dans la moitié cas, il s’agit d’un glioblastome d'emblée ou glioblastome primaire.
Dans les autres cas, il s’agit d’une progression d'un astrocytome de grade II ou II. On parle alors de glioblastome secondaire.
L’âge de survenue est légèrement différent : entre 40 et 50 ans pour les glioblastomes secondaires et après 60 ans pour les glioblastome primaires.
Des anomalies génétiques différentes...
Pour les glioblastomes primitifs : une amplification du récepteur du facteur de croissance épidermique (EGFR) associée à une perte d'hétérozysité du chromosome 10q, une délétion du PTEN (Phosphatase and tensin homologue on chromosome TEN) , une délétion affectant le chromosome 16 (p16).
Pour les glioblastomes secondaires : une inactivation successive de gènes suppresseurs de tumeur, P53, une surexpression du récepteur du facteur de croissance dérivé des plaquettes (PDGFR), des anomalies des gènes P16/CDKN/2A , ou le PTEN/MMAC1 du chromosome 10 ainsi que des pertes affectant les chromosomes 1p et 19q, dans les oligodendrogliomes de haut grade
La progression d’un phénotype peu agressif vers une tumeur hautement maligne résulte de l’accumulation de ces différentes altérations. Leur combinaison et l’ordre dans lequel elles surviennent permettent de dresser la carte d’identité moléculaire des tumeurs et de tracer des voies de tumorigenèse et de progression tumorale, comme l’illustre le tableau ci-dessous emprunté au site Internet de l’Association de Recherche sur les Tumeurs Cérébrales ( ARTC ).
LES OLIGODENDROGLIOMES
EN BREF...
Ils représentent environ 5 %des gliomes de l'adulte.
Ces tumeurs ont pour origine les cellules qui fabriquent la myéline (couche graisseuse qui protège les nerfs), les oligodendrocytes.
En général, ces tumeurs naissent dans le cerveau.
Elles affectent, le plus souvent les adultes d'âge moyen mais ont été décelées aussi chez des patients de tous âges.
Les spécialistes ont montré que ces tumeurs présentaient une mutation d’IDH1 ou d’IDH2 et d’une co-délétion 1p/19q et étaient dépourvues de signes d’agressivité histologique
La tumeur, habituellement bien limitée, est le siège de calcifications et est fréquemment localisée au lobe frontal.
Elle se traduit par les signes communs aux tumeurs : épilepsie, paralysies, déficits sensitifs, hypertension crânienne (maux de tête, vomissements).
Les oligodendrogliomes, dans 90 % des cas, présentent des calcifications périphériques. L’œdème péri-lésionnel est généralement minime.
CE QU'IL FAUT RETENIR...
Elles se développent lentement et ne s'étendent, généralement, pas aux tissus cérébraux avoisinants. En l’absence de traitement, après une durée variable, en général de plusieurs années, la plupart des cas évoluent vers des formes agressives anaplasiques.
Il n'existe pas de traitement "standard" validé. Il existe parmi les neurochirurgiens, un consensus en faveur de la réalisation, quand elle possible, d’une chirurgie aussi large que possible. Chez les patients qui ne peuvent être opérés, une biopsie est nécessaire au diagnostic et souvent un traitement complémentaire par le temozolomide ou une thérapie ciblée sera proposée.
À grade de malignité équivalent, ces tumeurs sont de meilleur pronostic que les tumeurs astrocytaires.
LE MEDULLOBLASTOME
SURTOUT UNE MALADIE DE L'ENFANT
C'est l'une des tumeurs malignes cérébrales la plus fréquente chez l’enfant, représentant, selon les études de 13 à 28 % des tumeurs cérébrales et 4 % de l'ensemble des tumeurs cérébrales.
On compte 50 nouveaux cas chaque année avec un pic entre 3 et 8 ans et une prépondérance masculine. Ils sont exceptionnels chez l’adulte, car 75 % des médulloblastome concerne des enfants.
La plupart des médulloblastomes se développent dans le cervelet; cependant, ils peuvent se produire également dans d'autres parties du cerveau. Ils dérivent vraisemblablement de cellules immatures de la couche des grains. Leur profil d’altérations génétiques est encore mal connu.
EN DEUX MOTS ...
Les symptômes fréquents
Ce sont des maux de tête, des vomissements dus à l'hydrocéphalie. Elle est due à un excès de LCS.
Des problèmes visuels (diplopie intermittente ou permanente), des problèmes d'écriture, une baisse scolaire, de l'indifférence, de l'apathie peuvent être aussi rencontrés. Le diagnostic a lieu habituellement de un à trois mois après le début des symptômes.
Son traitement
Il s'est beaucoup amélioré au cours des 20 dernières années.
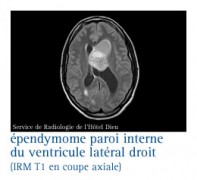
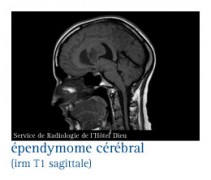
LES ÉPENDYMOMES
LEURS CARACTERISTIQUES
Ce sont des tumeurs se développent, le plus souvent, dans la paroi interne des ventricules cérébraux. Le siège fréquent est le quatrième ventricule (sous-tentoriel). Ils peuvent aussi naître dans la moelle épinière et le filum terminal ou cône médullaire.
La classification OMS 2007 retient 3 grades :
- Grade I : subépendymome (tumeur bénigne, d’évolution lente, attachée aux parois des ventricules) et épendymome myxopapillaire (siège dans la région du cône médullaire)
- Grade II :
- Grade III : épendymome anaplasique.
Bien que ces tumeurs puissent se développer à tout âge, elles sont plus fréquentes au cours de l'enfance et de l'adolescence (10 % des tumeurs cérébrales de l’enfant).
Ils entraînent un blocage de la circulation du liquide céphalo-spinal (LCS) qui provoque le développement d’une hypertension crânienne. Elle se traduit par des maux de tête non calmés par les médicaments usuels et, plus tard, des vomissements. Ce blocage du LCS peut aboutir à une hydrocéphalie.
TRAITEMENT
Les traitements actuels font appel à la neurochirurgie qui doit faire une exérèse de la tumeur la plus complète possible, en une ou plusieurs interventions, si nécessaire.
La radiothérapie focale reste le traitement standard chez l’enfant. Le rôle de la chimiothérapie fait l'objet d'études.
Chez le nourrisson, la chimiothérapie est toujours proposée en première intention.
En cas de récidive, une nouvelle chirurgie et la radiothérapie sont des options. L’inclusion dans des essais thérapeutiques est encouragée.
EN RÉSUMÉ…
|
Type de tumeur |
Cellule d’origine |
Fonction de la cellule |
|
Gliomes |
|
|
|
Oligodendrogliomes & |
|
|
|
Épendymome |
|
|
|
Médulloblastome |
|
|
POUR VOUS AIDER...
@ Association Michel Esnault : GFME Bât A, boîte 4, 22 Bd Camille Flammarion 13001 Marseille Tél : 04 91 64 55 86 - Courriel : gfme@free.fr
Mise à jour
22 novembre 2021