
Leucémies aiguës myéloïdes (LAM)
De l’adulte dans plus de 80 % des cas
EN BREF...
Les leucémies aiguës myéloïdes (LAM) représentent un ensemble très hétérogène de proliférations clonales de progéniteurs hématopoïétiques myéloïdes (blastes) au sein de le moelle osseuse, responsables de l’inhibition de l’hématopoïèse physiologique.
L’âge médian de découverte de la maladie se situe autour de 67 ans.
L’incidence annuelle, en France, est estimée à 3 à 4 cas pour 100 000. Elle est en augmentation pour des raisons obscures.
UN BLOCAGE DE LA MATURATION
Elles se développent à partir d’une cellule souche ou d’un progéniteur myéloïde ayant des caractéristiques d’auto-renouvellement, bloqué dans sa différenciation. La conséquence de cette prolifération massive et de ce blocage font que les précurseurs myéloïdes ne mûrissent pas et s’accumulent dans la moelle osseuse ou dans d'autres organes car ils ne peuvent pas mourir par apoptose. Ils s'immortalisent... .
UN HIATUS
On parle de hiatus leucémique car, dans la pyramide de maturation, il manque certains précurseurs. Cette discontinuité dans les différentes formes jeunes est caractéristique de ces maladies. Ceci explique le passage, dans le sang périphérique, de cellules immatures.
Les mutations à l'origine de la prolifération et du blocage de la différenciation...
| Mutations aboutissant à une prolifération cellulaire | Mutations bloquant la différenciation |
|---|---|
| FLT3 KIT RAS PTPN11 JAK2 |
PML-RARA RUNX1-RUNX1T1 CBFB-MYH11 MLL fusions CEBPA |
Les classifications
LA CLASSIFICATION FAB
Selon le niveau de blocage des lignées cellulaires de la moelle osseuse, la classification Franco-Américano-Britannique (FAB) distingue 9 types différents de LAM. Le chiffre entre parenthèse, donne l’incidence de la forme parmi les LAM et la formule de translocation :
- LAM0 : leucémie aiguë myéloblastique avec maturation minimale (3 %)
- LAM1 : leucémie aiguë myéloblastique sans maturation (15 à 20 %) ; t(9;22)
- LAM2 : leucémie aiguë myéloblastique avec maturation (25 à 30 %) ; t(8;21)
- LAM3 : leucémie aiguë promyélocytaire ou APL (5 à 10 %) ; (t15;17) associée au gène de fusion PML-RARα
- LAM4 : leucémie aiguë myélomonocytaire (20 %)
- LAMA4Eo : avec éosinophiles anormaux (5 à 10 %)
- LAM5 : leucémie aiguë monocytaire (2 à 9 %) ; (t9;11)
- LAM6 : érythro-leucémie aiguë (3 à 5 %)
- LAM7 : leucémie aiguë mégacaryocytaire (3 à 12 %)
LA CLASSIFICATION OMS DE 2008
À la classification FAB, morphologique, a succédé la classification de l’OMS qui fait intervenir d’autres caractéristiques, notamment les anomalies cytogénétiques.
1) LAM avec anomalies cytogénétiques récurrentes
a. Translocations équilibrées / inversions
- LAM avec t(8;21) (q22;q22) ; RUNX1-RUNX1T1
- LAM avec t(15;17) (q22;q12) ; PML-RARA
- LAM avec inv(16)(p13.1q22) ou t(16;16)(p13.1;q22) ; CBFB-MYH11
- LAM avec t(9;11)(p22;q23) ; MLLT3-MLL
- LAM avec t(6;9)(p23;q34) ; DEK-NUP214
- LAM avec inv(3)(q21q26.2) ou t(3;3)(q21;q26.2) ; RPN1-EVI1
- LAM (mégacaryoblastique) avec t(1;22)(p13;q13) ; RBM15-MKL1
b. Mutations génétiques
- Favorables : NPM1 muté isolé sans FLT3, CEBP alpha
- Défavorables : FLT3, KIT
2) LAM avec anomalies associées aux myélodysplasies
- Faisant suite à un syndrome myélodysplasique ou un syndrome myéloprolifératif / dysplasique : soit présentant des anomalies cytogénétiques identiques à celles des myélodysplasies, soit présentant une dysplasie sur > 50 % des cellules d’au moins 2 lignées myéloïdes
- Absence de radiothérapie ou chimiothérapie antérieure
- Pas d’anomalies cytogénétiques récurrentes
3) LAM post-chimio ou radiothérapie
Une seule entité, quel que soit le traitement
4) LAM sans spécification particulière
Reprend la classification FAB (M0 à M7 hors M3), en l’absence d’anomalies cytogénétiques ou moléculaires pouvant les classer ailleurs.
- + LAM basophile
- + Panmyélose avec myélofibrose
- LAM avec différenciation minime
- LAM sans maturation
- LAM avec maturation
- LA myélomonocytaire
- LA monoblastique / monocytaire
- LA érythroïde : LA érythroïde pure (érythro-leucémie)
- LA mégacaryoblastique
5) Sarcome granulocytaire
6) Proliférations myéloïdes associées à la trisomie 21 constitutionnelle
- Réaction leucémoïde transitoire
- LAM associée à la trisomie 21 constitutionnelle
Quelques données épidémiologiques...
Les données par sous-types issues des registres spécialisés, montrent que les LAM les plus fréquentes sont les LAM avec dysplasie multi-lignées.
Les LAM avec anomalies cytogénétiques ont un taux d’incidence annuel de 0,4/100 000 dont 60 % sont des LAM avec t(15 ;17).
Parmi les autres types de LAM, les LAM avec maturation sont les plus fréquentes avec un taux annuel d’incidence de 0,4/100 000 h.
La classification OMS de 2022 des LAM
|
Avec anomalies génétiques définies |
Définies selon le niveau de différenciation |
|
|
L’identification des anomalies chromosomiques
ESSENTIELLE POUR LE TRAITEMENT...
C’est une information très importante, à la fois sur le plan théorique mais aussi pronostique et thérapeutique car la connaissance d’une mutation précise peut déboucher sur des médicaments hautement spécifiques et plus actifs.
CONSIDÉRÉES COMME DE BON PRONOSTIC...
La translocation t(8;21)(q22;q22)
C’est la plus fréquente des translocations observées dans les LAM. Elle se retrouve chez la moitié des patients présentant une LAM2 mais se retrouve aussi dans d’autres leucémies aiguës. Elle représente près de 20 % des LAM de l'enfant.
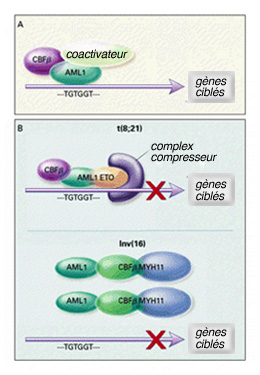
Il s’agit d’une translocation affectant les chromosomes 8 et 21. Une partie du bras long du chromosome 8 contenant le gène ETO est réciproquement transloquée sur le bras long du chromosome 21 au niveau du gène AML1 .
La conséquence de la translocation aboutit à un gène de fusion entre le gène AML-1 et le gène ETO dans sa partie CBFb. Le gène de fusion s’appelant AML1/CBFb n’est plus capable de réguler les fonctions cellulaires. Ces anomalies sont représentées sur le schéma.
Le pronostic des LAM avec t(8;21) est dans l’ensemble bon, à la fois en termes de rémissions avec un taux proche de 85 % et qu'en termes de survie, proche de 60 %, en particulier avec l'utilisation de fortes doses d'aracytine.
La translocation t(15;17)(q25;q21)
L'anomalie caractéristique de la leucémie à promyélocytes (LAP ou LAM3). consiste en une translocation entre le chromosome 15 et le chromosome 17.
La translocation englobe le récepteur de l’acide rétinoïque alpha (RAR-alpha) du chromosome 17 et la protéine PML (Promyelocytic Leukemia Protein), codée par le chromosome 15. Il n’est pas encore bien élucidé comment la protéine de fusion aberrante PML-RAR- alpha conduit à la leucémie.
Les récepteurs à l’acide rétinoïque sont des facteurs de transcription qui affectent de nombreux processus physiologiques, dont l’hématopoïèse. Ils exercent leurs effets biologiques en se liant sous forme à des éléments spécifiques régulateurs appelés RAREs pour Retinoic Acid Response Elements .
Le transcrit PML-RAR-alpha étant spécifique des LAP, sa recherche est maintenant systématique car il existe des traitements spécifiques, ATRA (Acide tout transrétinoïque) d’une part et un dérivé de l'arsenic, le Trisinox™, d’autre part. II s'agit du premier exemple de traitement spécifique d'une leucémie aiguë.
Depuis, l’introduction de ces nouvelles thérapeutiques, le taux de rémission complète est aujourd'hui proche de 100 %, avec un taux de survie proche de 80 % en dépit d'un risque hémorragique.
La translocation inv(16)(p13q22)
Cette anomalie est caractéristique de la LAM4 à éosinophiles. Elle englobe le gène MYH11 (MYosin Heavy chain) et le gène CBFb (facteur de transcription de la cellule T) du chromosome 16.
Cette leucémie à un bon pronostic.
LES AUTRES DE PRONOSTIC VARIABLE
La translocation t(9;22)(q34;q11)
Elle se voit, le plus souvent, dans la LAM1 et dans la LAM2.
La modification est similaire à celle observée la leucémie myéloïde chronique. Elle aboutit au gène BCR -ABL à l’origine de la production de la protéine bcr-abl responsable de la prolifération du clone cellulaire malade.
La translocation t(6;9)(p23;q34)
Elle est peu spécifique est implique les gènes DEK et CAN .
Les leucémies, avec cette translocation, sont de moins bon pronostic.
Le réarrangement en 3q21
Il est généralement associé à une augmentation du nombre de plaquettes (thrombocytose).
Le réarrangement en 11q23 du chromosome 11
Il est observé en cas de LAM4 et de LAM5 qui affecte les âges extrêmes de la vie. De plus, c'est dans 80 % des cas, la forme associée aux leucémies secondaires, après exposition à des médicaments bloquant les topoisomérases II, comme les anthracyclines.
Les éléments pronostiques
CERTAINES CARACTÉRISTIQUES DE LA MALADIE
De nombreuses recherches ont permis d'établir qu'un certain de nombre de facteurs permettaient d'estimer le pronostic de la maladie et d'optimiser la stratégie thérapeutique à mettre en œuvre.
Les facteurs suivants ont été validés et sont maintenant utilisés, en routine pour établir le pronostic de la leucémie.
- L'âge : moins de 60 ans versus plus de 60 ans
- Le caractère primaire ou secondaire de la leucémie
- Une augmentation du nombre de globules blancs (hyperleucocytose) initiale supérieure à 30 x 10 9 /l
- Le type de cellules proliférant (type cytologique) : meilleur pronostic pour les formes M3 et M4 à éosinophiles ; moins bon pronostic des formes M0 et M7
- Le phénotype immunologique : pronostic réservé en cas d’expression à la surface des cellules tumorales de la protéine CD34+ et/ou de la protéine gp170 codant le gène de résistance multiple aux médicaments, MDR1 (Multiple Drug Resistance)
LE PHÉNOTYPE DU CLONE TUMORAL
Les anomalies cytogénétiques propres au clone de cellules tumorales proliférant sont mises en évidence par l'analyse du caryotype. Le profile cytogénétique est réalisé à partir de l'étude des chromosomes des cellules blastiques en métaphase, lors de leur division cellulaire.
Des études récentes ont proposé d’incorporer dans l’arbre décisionnel thérapeutique, une évaluation cytogénétique comprenant aussi les gènes FLT3, NPM1, CEBPA et KIT voire, DNMT3A, IDH1/2 et TET2 .
C'est l'information la plus pertinente pour estimer le pronostic. Certaines translocations sont de bon pronostic et une bonne réponse aux chimiothérapies de consolidation, sauf s’il s’agit de LAM secondaires :
- La translocation t(15;17) de la LAM 3 et créant un gène de fusion entre le gène PML et le gène RARA codant pour le récepteur nucléaire à l’acide rétinoïque
- La translocation t(8;21) retrouvée dans environ 25 % des LAM 2 et créant un gène de fusion AMLI/ETO caractéristique de la LAM 4 avec précurseurs éosinophiles médullaires anormaux et créant un gène de fusion CBFB/MYH11
- La translocation RUNX1-RUNX1T1.
Les autres anomalies sont plutôt de moins bon pronostic et sont plutôt redevables d’une greffe allogénique de moelle osseuse. Il s'agit, en particulier
- Des trisomies du chromosome 8
- Des anomalies des chromosomes 5 et/ou 7 ou du chromosome 11 [(bande (11q23)]
- De l'inversion du chromosome 13:inv(3)
- Des remaniements chromosomiques complexes qui sont associés aux transformations aiguës de certains syndromes myélodysplasiques, comme l'anémie réfractaire avec excès de blastes (AREB) et aux LAM secondaires
Les formes dont les cellules ont des caryotypes normaux sont considérées comme ayant un pronostic intermédiaire. La réponse aux traitement est alors variable.
Rarement il s'agit de formes bi-phénotypiques, c'est-à-dire ayant le phénotype myéloblastique et lymphoblastique. Ce sont des formes très immatures, dont les cellules tumorales, les blastes, portent à la fois des marqueurs de différenciation myéloïdes et lymphoïdes précoces. Le pronostic de ces formes très rares est réservé.
La nouvelle classification pronostique selon l’ELN de 2022
|
Pronostic |
Anomalies génétiques |
|---|---|
|
Favorable |
|
|
Intermédiaire |
|
|
Défavorable |
|
Mise à jour
16 juin 2023