
Leucémies aiguës lymphoblastiques (LAL)
Dans 80 % des cas, ce sont des enfants ...
DES PATHOLOGIES PRINCIPALEMENT "B"
Dans les trois quarts des cas, ce sont des proliférations massives de précurseurs des lymphocytes B et dans moins d’un quart des cas des lymphocytes T.
Ces cellules sont identifiées grâce à l'immunophénotypage qui permet de mettre en évidence des protéines spécifiques de surface appelées CD (Cluster of differenciation) ou des antigènes particuliers, les immunoglobulines. Ces techniques permettent de reconnaître la lignée impliquée et le degré de maturation de la cellule maligne.
Chez l'adulte, dans un quart des cas on retrouve un chromosome Philadelphie comme dans les leucémies myéloïdes chroniques.
DES PROGRÈS ÉVIDENTS ...
Les traitements modernes (induction, consolidation et maintenance) ont permis de passer, chez l'enfant d'un taux de guérisons de 40 %, en 1960, à plus de 80 %, actuellement. Néanmoins, 15 à 20 % des enfants rechuteront et nécessiteront une nouvelle chimiothérapie dite "de rattrapage" intensive et une allogreffe de cellules souches hématopoïétiques dans la majorité des cas. Ces traitements intensifs permettront de sauver 30 à 50 % de ces enfants.
Chez l'adulte, les chiffres sont moins spectaculaires avec moins de 20 %, en 1960 à plus de 60 % de survie à 5 ans, en 2015.
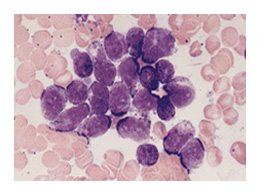
Les classifications
LA CLASSIFICATION FAB
La cytologie est à la base de la classification Franco-Américano-Britannique FAB qui distingue trois grands types de LAL
- LAL1: leucémie aiguë lymphoblastique avec une population cellulaire homogène avec > 75% de petites cellules
- LAL2: leucémie aiguë lymphoblastique avec une population cellulaire hétérogène avec des cellules, en moyenne, larges
- LAL3 : leucémie aiguë lymphoblastique avec une population homogène à grandes cellules ressemblant aux lymphoblastes du lymphome de Burkitt.
LA CLASSIFICATION DE L'OMS DE 2016
Le principe : la fusion entre les leucémies et les lymphomes
Dans cette classification, les leucémies aiguës lymphoblastiques sont classée en tant que cancers affectant les précurseurs lymphoïdes B et T.
Elle regroupe lymphomes et leucémies, considérant que si la présentation au diagnostic varie, c'est le même précurseur qui est en cause. C'est le cas de la cellule impliquée dans la tumeur de Burkitt dont l'expression morphologique est tantôt une masse solide, c'est-à-dire un lymphome de Burkitt, tantôt une leucémie, correspondant à la LAL3 de la classification FAB.
Selon le point de départ de la tumeur, la présentation et les signes de la maladie vont changer.
- Si l’événement aboutissant à la transformation maligne débute dans la moelle osseuse produisant les cellules du sang (hématogène), la maladie se présentera sous l’aspect d’une leucémie lymphoblastique B, en raison du passage rapide des cellules tumorales dans la circulation sanguine, les adénopathies seront au second plan.
- Si, l’événement initial de la tumeur a lieu dans un organe ou une localisation viscérale, l’expression clinique de la maladie sera celle d’une tumeur solide. La maladie sera donc considérée comme un lymphome.
Les leucémies lymphoblastiques des précurseurs B / lymphome lymphoblastique (LAL-B)
Elles correspondent aux formes L1 et L2 de la classification FAB. Dans trois quarts des cas, ce sont des maladies de l’enfant et dans près de 85 % des cas, elles sont de phénotype B . Les causes précises de ces maladies sont inconnues, à ce jour. Les principales anomalies rencontrées sont les suivantes :
- Leucémie aiguë / lymphome lymphoblastique B sans autre spécification
- Leucémie aiguë / lymphome lymphoblastique B avec anomalies cytogénétiques récurrentes avec :
- t(9;22) (q34;q11.2) ; BCR-ABL1
- t(v;11q23) ; MLL réarrangé
- t(12;21)(p13;q22) ; TEL-AML1 (ETV6-RUNX1)
- Hyperploïdie
- Hypodiploïdie
- t(5;14)(q31;q32) ; IL3-IGH
- t(1;19)(q23;p13.3) ; TCF3 - PBX1
Les différentes formes sont présentées plus en détails dans le chapitre suivant.
Les leucémies lymphoblastiques des précurseurs lymphoblastiques T / lymphome lymphoblastique T
Elles sont moins fréquentes
Elles ne représentent que 15 % des LAL de l’enfant et 25 % des LAL de l’adulte. Elles sont caractérisées par une leucocytose élevée et un syndrome tumoral important, en particulier thymique.
Les anomalies génétiques rencontrées dans les LAL-T
Elles sont nombreuses. Elles impliquent en particulier les gènes du récepteur T à l’antigène (TCR) dans 30 % des cas, dont les régions régulatrices, fortement activées au cours de la différenciation T dans le thymus, sont rapprochées de différents gènes partenaires. Ce rapprochement physique conduit à l’activation du gène partenaire, à un moment imprévu et dans un site aberrant, créant l’événement aboutissant à la prolifération tumorale.
Dans les translocations impliquant le récepteur TCR, il n’y a pas de formation de transcrit de fusion.
Les gènes les plus impliqués
Ce sont, le MYC , le TAL , le RBTN1 et 2 , le HOX11 et le LCK .
Dans d’autres cas, on retrouve des délétions du chromosome 9p, responsable de la perte du gène suppresseur de tumeur CDKN2A ou des micro-délétions à l’intérieur du gène TAL1 , responsable de l’expression anormale de ce facteur de transcription.
En pratique, seules la microdélétion de TAL1 , l’expression de HOX11 et de HOX11L2 sont recherchées pour affiner le diagnostic.
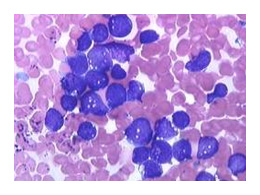
Les anomalies chromosomiques
DEUX TYPES DE TRANSLOCAION LES PLUS COMMUNES
Deux types fonctionnels de translocations sont habituellement retrouvées.
- Les oncogènes sont relocalisés dans les régions régulatrices des gènes activement transcrits, causant une dérégulation de l’expression d'une protéine normale.
- La translocation juxtapose deux gènes qui codent pour une protéine chimérique qui possède des fonctions distinctes des protéines normalement transcrites par ces gènes.
LES LEUCEMIES DE LA LIGNÉE LYMPHOCYTAIRE B
ETV6-RUNX1 (TEL-AML1) = translocation t(12;21) (p 13;q22)
C'est la plus fréquente des translocations retrouvées dans les LAL B2 ou commune.
On observe une translocation t(12;21 ) dans environ 25 % des LAL de l'enfant contre moins de 3 % chez l’adulte.
C’est une translocation aboutissant au gène de fusion TEL-AML1 . Il est généré par la translocation t(12;21)(p13;q22). Ce gène de fusion est rencontré dans 25 % des leucémies lymphoblastiques à cellules B.
Cette mutation peut être déjà présente dans le sang du cordon ombilical et conférer aux cellules souches mutées de se renouveler et d’inhiber les phénomènes d’apoptose.
Le pronostic de cette forme de la maladie est bon.
Toutefois, si le risque de rechute n'est que légèrement plus faible que dans les autres LAL, ces rechutes surviennent en général de façon plus tardive et sont donc moins graves.
PBX-E2A = translocation t(1;19)(q22;p13.3)
Elle juxtapose la région 5' du gène E2A , sur le chromosome 19, et les régions 3' de PBXl sur le chromosome 1. Elle aboutit à la création du gène de fusion E2A-PBX1 .
Le pronostic des LAL associées à cette translocation était autrefois peu favorable. Depuis l’instauration des intensifications thérapeutique, le caractère péjoratif de cette anomalie semble avoir disparu.
Le réarrangement du gène c-MYC
La translocation t(8;14)(q24;q32) et ses variantes sont caractéristiques de la LAL3 et du lymphome de Burkitt (cellule maligne B mature).
Le chromosome 8, en 8q24, porte l'oncogène c -MYC et le chromosome 14, en 14q32, commande la production des chaînes lourdes d'immunoglobuline. La translocation met l'oncogène sous la dépendance d'un promoteur de chaîne d'immunoglobuline et son expression est alors très augmentée.
Initialement considéré comme péjorative, son pronostic s'est amélioré avec la technique d'intensification thérapeutique.
AF4-MLL = translocation t(4;11)(q21;q23)
C’est une translocation impliquant le gène MLL (11q23) du chromosome 11 et le gène AF4 (4q21) du chromosome 4. Le gène de fusion conduit à la production d’une cellule B immature (CD19+).
Elle touche l'enfant très jeune et peut même aboutir, parfois, à une leucémie congénitale, c’est-à-dire se développant avant l'âge d'un an.
Le seul traitement efficace de cette leucémie est la greffe de moelle.
BCR-ABL = translocation t(9;22)(q34;q11)
C’est une translocation impliquant le gène ABL et le gène BCR. Elle aboutit, comme dans les LAM ou les LMC, à la production d’une protéine mutée à activité tyrosine kinase soit p210 soit p190 dans l'autre moitié des cas.
Le traitement de cette forme de leucémie est plus difficile.
DE LA LIGNÉE LYMPHOCYTAIRE T
Les réarrangements du gène TAL-1
Les remaniements du gène TAL-1 , localisé en 1 p32, sont retrouves dans 20 à 30 % des LAL-T de l'enfant, dont ils constituent une des anomalies les plus fréquentes.
Deux types de réarrangement aboutissent à la surexpression de TAL-1. La signification pronostique est mal connue.
La translocation t(5;14)(q35;q32) et surexpression du gène HOX11L2
Récemment, les analyses par FISH ont permis d'identifier cette nouvelle translocation. Elle est retrouvée dans 25 % des LAL de la lignée T de l’enfant. Cette anomalie implique de manière constante le gène Ran BP17 , localisé en 5q35. Cette translocation induit une surexpression du gène HOXl1L2 , localisé, lui aussi, en 5q35.
La surexpression du gène HOXl1L2 est de pronostic défavorable.
Bien d'autres translocations connues ou à découvrir...
Elles sont très nombreuses et beaucoup restent à découvrir. A titre d’exemple, on peut citer les translocations t(11;14)(p13;q11), t(8;14)(q24;q11) et t(10;14)(q24;q11).
Les facteurs pronostiques reconnus
AVANT TRAITEMENT
Les marqueurs cytogénétiques
De bon pronostic : absence d’anomalies, les hyperploïdie à plus de 50 chromosomes et/ou une translocation t(12;21).
De pronostic réservé : les anomalies de nombre, la translocation t(9;22)
L'âge
Pronostic meilleur entre 2 et 10 ans, pronostic moins bon avant l'âge d'un an
L'importance de la masse tumorale
Pronostic réservé en cas de syndrome tumoral clinique, d'une masse médiastinale, d'un envahissement méningé initial, une élévation du taux de la LDH
Le type cytologique
LAL commune origine pré B, porteuse de l'antigène CD10+ ou Calla de bon pronostic ; L3 défavorable, à cause du syndrome de lyse et LAL T hyperleucocytaires.
Les marqueurs immunologiques
Pronostic réservé si présence à la fois de marqueurs des lymphocytes B et de marqueurs des lymphocytes T ; meilleur pronostic pour les LAL BII.
LA RÉPONSE AU TRAITEMENT
Les principaux facteurs pronostiques sont la réponse précoce à la chimiothérapie, notamment aux corticoïdes chez l'enfant et l'évolution de la maladie résiduelle évaluée par la méthode de la PCR utilisant les réarrangements des chaînes d'immunoglobulines (LAL de type B) ou des récepteurs T (LAL de type T et souvent B). Sont de bon pronostic, une maladie résiduelle <1 % après le traitement d'induction, et <1 ‰, après le traitement de consolidation.
Deux hypothèses pour expliquer la maladie
La première de Greaves
Elle est basée sur l'observation que les événements initiateurs de la LAL infantile.
Le « first hit de Knudson », survient in utero à partir de l'expansion clonale d'un groupe de cellules hématopoïétiques porteuses de mutations mais n'ayant pas progressé jusqu'à un stade de leucémique ce sont des clones pré-leucémiques.
Au sein d'un sous-groupe d'individus qui hébergent des clones pré-leucémiques du « first hit », l'absence d'exposition aux infections dans la petite enfance, combinée à une anomalie génétique ultérieure déclenchée après la naissance en raison d'une infection induite par le stress (second hit), interagit avec le système immunitaire en développement dans la petite enfance.
Cette interaction complexe, connue sous le nom d'hypothèse de « l'infection retardée » de Greaves, favorise potentiellement la LAL 37 chez l'enfant
La deuxième hypothèse dite de Kinlen
Elle est basée sur une approche épidémiologique qui cherche à expliquer la pathologie au travers des clusters épidémiques. Connue sous le nom d'hypothèse du « mélange de population » d'après les travaux de Kinlen et ses collègues. Dans ce scénario, la LAL est considérée comme le résultat de l'exposition à de nouveaux agents pathogènes résultant d'un mélange accru de la population. Cet effet est particulièrement évident lorsqu'un grand nombre d'individus, y compris de nombreuses personnes issues de milieux urbains qui sont par conséquent exposés à diverses infections, s'installent dans une zone peu peuplée où une partie importante de la population locale n'a pas été exposée auparavant à cet agent
Mise à jour
8 mars 2021